Active Studies
Home | Our People | Active Studies | Past Studies | Tools | Researchers | Participants
Family Based Analyses to Identify Rare Genetic Variants
Platelet function assays are moderately to highly heritable supporting the hypothesis that genetic variations underlie individual variability in the tendency for arterial thrombosis. During the past seven years, in GeneSTAR GWAS revealed multiple common genetic loci that pass stringent GWAS thresholds in African American and European Americans families at high risk for CHD. Common variants were found to determine variability in native platelet aggregation as well as residual platelet aggregation after low dose aspirin (ASA) intervention. However, collectively the loci identified through this common variant approach account for less than 35% the total heritability of these phenotypes in the GeneSTAR families. Extending a family-based design in an integrative approach, our approach is currently : 1) identifying rare variants in genes that are associated with native and residual post- ASA platelet aggregation, testing the hypothesis that a significant fraction of the 'missing heritability' in platelet aggregation phenotypes is due to these rare variants; and 2) follow up -identified loci to determine the underlying 'causal' variants tagged by the GWAS association signal. In a family-based sequencing approach we have sequenced 200 hyper-aggregable individuals selected from African American and European American GeneSTAR families with clustering of platelet aggregation.
Validated exome sequencing-identified genes along with the GWAS-identified loci will be followed up relying on a targeted deep resequencing approach of 1,300 African American and European American subjects from additional GeneSTAR families. The results from this integrative GWAS and exome approach will lead to a better understanding of the role of genetic variants (common and rare) in the determination of platelet aggregation native and residual post-ASA, including possible racial differences, and should enable genotypic tailoring of preventive therapy for CHD in high-risk individuals. Native and residual post aspirin platelet hyper aggregation, a strong risk factor for ischemic syndromes, is moderately to highly heritable.
Our in families suggest a high degree 'missing heritability' (i.e. that not explained by the common GWAS signal detected). The primary hypothesis is that genes harboring rare genetic variants determining platelet aggregation account for a substantial fraction of missing trait heritability, and an integrative family-based approach of GWAS and exome-sequencing will be applied to test this hypothesis.
Working closely with the Biostatistics Department, we are honing in on methods that utilize the full capacity of family-based methods,and that we will extend to whole genome sequencing, providing integration of our different approaches to understanding platelet function. Transitioning these novel analytic models to whole genome data and the multiomic data we are collecting in other studies will provide a valuable adjunct to larger studies using discovery approached in Big Data efforts.
Whole Genome Sequencing; TOPMed
Trans-Omics for Precision Medicine (TOPMed) Program: NHLBI

A supplement to this work was approved for NHLBI Whole Genome Sequencing Project (NHLBI-WGS) in 1800 subjects to significantly boost the study design of the parent work in families with platelet function. The new TOPMed work involves two additional RNASeq projects on specific target tissue of platelets and iPS derived megakaryocytes. Our primary emphasis is on the genetics of platelet aggregation and our original WGS study design was a two-step approach to identifying genes harboring rare variants that determine platelet hyper-aggregation in families at high risk for CAD. We are also generating data in families that will be far reaching beyond the primary phenotypes using the extensive phenotyping available of the participating subjects from families identified at high risk over a 35 year period of the GeneSTAR Program. In TOPMed under Dr. Mathias’s leadership we are participating in work groups on atherosclerosis, hematologic traits, neurology and related traits, anthropometrics, diabetes, and many other traits.
Innovation of the Family Based Design
The application of the family based study design is multi-fold wherein we (1) identify families that have clustering of baseline hyper-aggregation and residual post-ASA hyper-aggregation; (2) whole genome sequence a set of families prioritized on the basis of both phenotypes; (3) replicate the identified genes/loci in additional independent families with measured phenotype; and (4) leverage WGS and transcript data to prioritize non-coding sequence identified variants based on exhaustive eQTL analysis. Our original study design is vastly improved in its innovation with points 3 and 4 above yielding a comprehensive multi-step approach which not only yields high statistical power for rare variant association signal, but also limits the false positive signal seen as a limitation of rare variant investigation in the case-control design. Finally this approach offers new insight into how much of the 'missing heritability' can be explained by rare variants in the context of true familial heritability in place of total phenotypic trait variability.
Principal Investigator
Rasika A. Mathias, ScD
Associate Professor, Medicine
Associate Professor, Epidemiology
Collaborators
Kai Kammers, PhD
Assistant Professor, Oncology Center, Biostatistics and Bioinformatics
Jeff Leek, PhD
Professor, Biostatistics and Oncology
Ingo Ruczinski, PhD
Professor. Biostatistics
Margaret A. Taub, PhD
Assistant Scientist, Biostatistics
Life After Linkage
Gene Transcripts and Proteomics in Families with Platelet Hyperaggregation
Platelet hyperaggregation is an important intermediate phenotype for myocardial infarction, acute coronary syndromes, and strokes. We have already discovered and replicated GWAS signals for platelet aggregation in two-generational families of premature coronary disease probands (GeneSTAR), European Americans and African Americans. Although platelet aggregation is highly heritable, all of the identified GWAS signals together explain only a small fraction of its variance among individuals. GWAS signals are located in introns and intergenic regions, so it is not clear how the variant is functionally related to the aggregation response. In this application we propose to discover new pathways regulating platelet aggregation by determining which genes are expressed in subjects with platelet hyperaggregation. By sequencing the entire platelet transcriptome we will identify changes in the amount or quality (e.g., splice variants) of mRNA transcripts that are associated with specific platelet hyperaggregation phenotypes. We are also using whole genome sequencing and RNA-seq to address our research questions.
Our aims are to: (1) use a unique family-based design to examine genes that are differentially expressed in white and African American subjects with platelet hyperaggregation compared to control subjects (as defined from prior studies), (2) leverage our prior GWAS to identify eQTLs associated with transcript expression to help prioritize transcripts/genes for further study, and (3) use quantitative mass spectrometry to determine whether changes in expression in hyperaggregating platelets are accurately reflected in corresponding changes in expressed proteins.
This study will produce a complete quantitative inventory of all gene transcripts present in platelets, as well as a complete eQTL map of genetic loci responsible for transcript expression specifically in platelets in both European and African Americans. We expect that our studies will identify previously unknown proteins and biological pathways responsible for platelet hyperaggregation, which may then serve as new therapeutic targets and ultimately more effective and specific approaches for inhibition of platelet function in the large number of people at risk for thrombotic vascular occlusions being treated with anti-platelet therapy.
The study uses sibling pairs and global controls and is operational from 2014-2019.
Consent Form
Principal Investigator
Lewis C. Becker, MD
Robert L. Levy Professor, Cardiology
Induced Pluripotent Stem Cells, Megakaryocytes, and Platelets

This is a new grant commencing in 2011. Below we outline the Phases. GeneSTAR's NextGen is a collaborative project with the Divisions of General Internal Medicine, Hematology, and Cardiology, The Institute for Cell Engineering, and the Johns Hopkins Genetics Core. The project has multiple Principal Investigators. Under the leadership of Drs. Lewis Becker from GeneSTAR and Linzhao Cheng from hematology, the study began with a 2 year laboratory phase, a portion of which was guided by the University of Tokyo (Dr. Hiro Nakauchi) and University of Kyoto (Dr. Koji Eto).
Platelets in the circulating blood mediate normal hemostasis, but may also initiate pathological arterial thrombosis that produce heart attacks and strokes. In our GeneSTAR GWAS study of native platelet and post-aspirin platelet function, we found many signals of genome-wide significance. The mechanism has remained largely undefined because most signals have occurred in introns or intergenic regions rather than in protein coding regions of known genes.
In addition, platelets are derived from megakaryocytes in the bone marrow, but themselves are anucleate with little residual mRNA. In this 3 phase study, we examine the functional genomics of these associations in order to define novel risk assessment paradigms and identify new therapeutic targets for cardiovascular and thrombotic disorders.
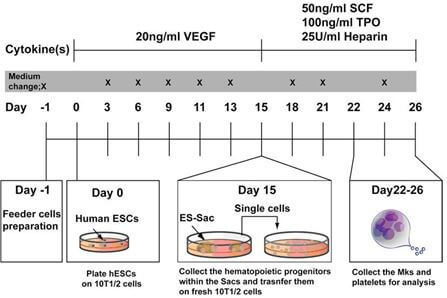
In Phase I, under the direction of Dr. Linzhao Cheng with assistance from Drs. Nakauchi and Eto,
- developed an efficient method to generate human pluripotent stem cells (iPS) from peripheral blood mononuclear cells,
- developed methods to generate differentiated megakaryocytes (Mks) from these human iPS,
- determined that these differentiated Mks look like normal Mks and possess the cell markers of naturally occurring Mks, and that the whole-genome genotype of these differentiated Mks remains “true” to the original genotype.
In Phase II, also under Dr. Cheng's direction,we:
- developed an efficient method to generate iPS cells in batches from at least 10 individuals at a time using PBMCs from 20 GeneSTAR subjects
- developed methods to generate Mks from these batches and performed RNAseq.
In Phase III, under the direction of the GeneSTAR team, Dr. Lewis Becker has
- generated iPS cells from 257 selected study subjects who have had whole genome sequencing, a GWAS and extensive platelet phenotyping, chosen by phenotype and/or genotype for SNP variants with genome-wide significant associations in white and African American subjects.
- Mks were differentiated from iPS cells for each subject selected. We examined gene expression profiling from the differentiated Mks from each subject using the Human Exon 1.0 ST Array from Affymetrix (containing all known gene transcripts and expressed sequence tags (ESTs) in humans), and confirmed the expression.
- The expression of selected proteins corresponding to the expressed mRNAs is being examined by mass spectrometry at the laboratory of Dr. Jenny Van Eyk, Cedars Sinai Research Institute, Los Angeles CA.
- For the significant genotype/phenotype SNP associations we have foundboth for native and post-ASA platelet function, we are comparing gene expression profiles from Mks by genotype.
- We are determining whether elevated transcripts are associated with expression of the corresponding proteins.
- We are determining the relationship between genetic variants across the genome and gene transcript levels (eQTLs) using multi-dimensional analysis to help understand how genetic variants, particularly in intergenic regions, may produce functional genomic effects. This will provide an eQTL database for megakaryocytes that is shared with the scientific community.
- We are also comparing gene expression profiles for Mks from subjects with high vs. low platelet aggregation response to different agonists (collagen, ADP, epinephrine, and arachidonic acid), at baseline and after aspirin.
- We are determining whether some expressed transcripts code for proteins in known functional pathways.
In Phase III, work includes collaborations of GeneSTAR's Dr. Rasika Mathias, an expert in genetic analyses, with Drs. Jeff Leek, Kai Kammers, and Margaret Taub in the Department of Biostatistics in the Bloomberg School of Public Health. We are also studying mutation rates in iPSC and copy number variation with the expertise of Dr. Ingo Ruczinski, Department of Biostatistics.
Cell Repository
We maintain an iPSC banking repository for all 257 cell lines at Johns Hopkins in the Becker Laboratory in Cardiology.
The cells are also available at WiCell, Inc. the home of the entire NHLBI NextGEN Project. WiCell can be searched using the following link to the Becker cells from GeneSTAR: https://www.wicell.org/home/stem-cells/catalog-of-stem-cell-lines/collections/nhlbi-next-gen-becker.cmsx
WiCell Repository
Platelet Aggregation (Dr. Lewis Becker, The Johns Hopkins University)
Becker Lab’s Next Gen Cell Lines This collection, from Dr. Lewis Becker (The Johns Hopkins University), was generated to enable the study of the genetic basis of human variation in native platelet function and platelet responsiveness to aspirin. The iPS cell lines from this study are included within this collection.
This collection contains 198 human induced pluripotent stem cell lines derived under highly efficient clinically compliant conditions. The cell lines are comprised of healthy brothers, sisters, and offspring of index cases and siblings of persons of early onset coronary disease (< 60 years of age). None of the subjects were affected with clinical coronary disease, stroke, or other overt vascular disease phenotypes at the time of the study. Pedigrees are detailed in the “Genetically Related Cell Lines” field of each cell line’s web page. The ages of donors range from 28 to 86 and ethnicities include European American and African American.
About the Next Generation Genetic Association Studies (Next Gen) Program
These cell lines were created as Next Generation Genetic Association Studies (Next Gen) Program, which was a five-year, $80 million program to investigate functional genetic variation in humans by assessing cellular profiles that are surrogates for disease phenotypes. To achieve this, researchers from multiple institutions across the U.S. were awarded grants to derive iPS cell lines from more than 1,500 individuals representing various conditions as well as healthy controls for use in functional genomic (‘disease in a dish’) research. This extensive panel includes a diverse set of age, gender and ethnic backgrounds, and therefore will be an invaluable tool for evaluations across demographics. Further enhancing the utility of these cell lines are data sets such as phenotyping, GWAS, genome sequencing, gene expression and -omics analyses (e.g., lipidomic, proteomic, methylomic) that will be made available with the cell lines.
iPS Megakaryocytes, and Platelets in GeneSTAR
Principal Investigator
Lewis C. Becker, MD
Robert L. Levy Professor, Cardiology