Celebrating 250 Years
Learn More
-
Medical Services and Care
Learn about our medical services provided by our team of experts.

-
Patients and Visitors
Learn how we strive to keep you comfortable during your stay with us.

-
Maps and Directions
4940 Eastern Avenue Baltimore, Maryland 21224.

-
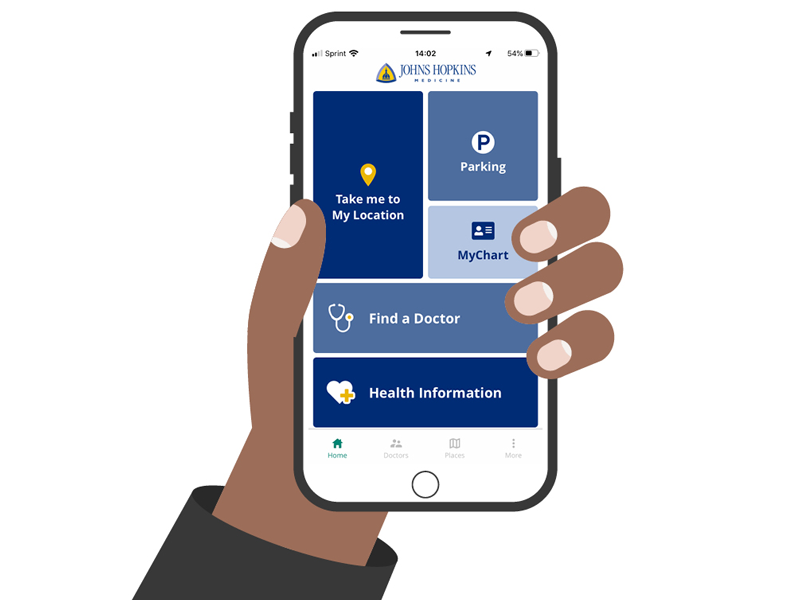
MyChart
Schedule appointments, view test results and more.



The Many Ways We Care for You
Learn how different teams at Johns Hopkins Bayview make sure you have a great experience as a patient. Throughout your visit, you will experience the capable, nurturing care that sets us apart.


