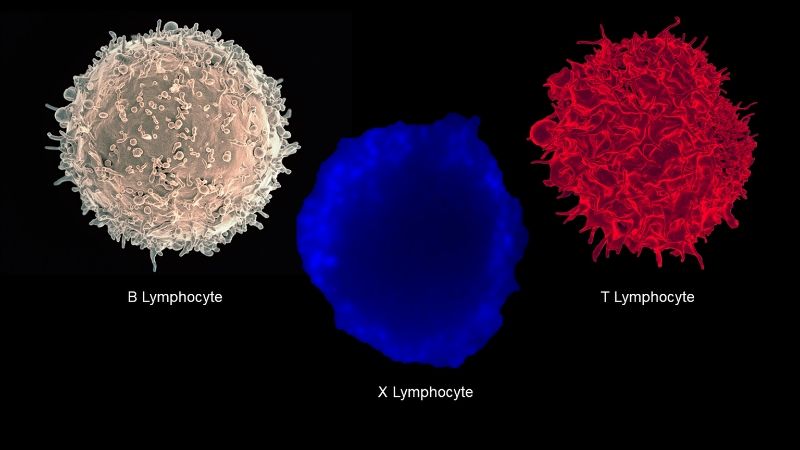
In a discovery that might be likened to finding medicine’s version of the Loch Ness monster, a research team from Johns Hopkins Medicine, IBM Research and four collaborating institutions is the first to document the existence of the long-doubted “X cell,” a “rogue hybrid” immune system cell that may play a key role in the development of type 1 diabetes.
The researchers report the unusual lymphocyte (a type of white blood cell) — formally known as a dual expressor, or DE, cell — in a new paper published in the journal Cell.
“The cell we have identified is a hybrid between the two primary workhorses of the adaptive immune system, B lymphocytes and T lymphocytes,” says Abdel-Rahim A. Hamad, M.V.Sc., Ph.D., associate professor of pathology at the Johns Hopkins University School of Medicine and one of the authors of the paper. “Our findings not only show that the X cell exists, but that there is strong evidence for it being a major driver of the autoimmune response believed to cause type 1 diabetes.”
Type 1 diabetes, formerly known as juvenile diabetes or insulin-dependent diabetes, is a chronic condition in which there is destruction of the beta cells in the pancreas that produce insulin, the hormone that regulates a person’s blood sugar level. Diagnosed mostly in childhood but present at all ages, the disease accounts for between 5% and 10 % of all diabetes cases in the United States or about 1.3 million people. Although most experts believe it to be an autoimmune disorder — where the immune system mistakes normal, healthy beta cells as hazards and eliminates them — the underlying mechanism at the cellular level has been difficult to define.
Hamad and his colleagues believe that they may be the first to do so. However, they caution that more analysis is required to directly link the X cell to the development of type 1 diabetes.
“What is unique about the entity we found is that it can act as both a B cell and a T cell,” Hamad says. “This probably accentuates the autoimmune response because one lymphocyte is simultaneously performing the functions that normally require the concerted actions of two.”
B and T lymphocytes each possess distinctly different cell receptors — the B cell receptor, or BCR, and T cell receptor, or TCR, respectively — that work together to help identify and target antigens; the bacteria, viruses and other foreign invaders that trigger an immune response. Normally, this defense begins when the trespasser is engulfed by a white blood cell called an antigen presenting cell, or APC. The name arises from the fact that an antigenic protein from the ingested intruder is “presented” on the surface of the APC.
After this occurs, the APC travels to a part of the body, such as a lymph node, where immature B and T cells reside. A T cell with a TCR whose shape conforms to the presented antigen — akin to fitting a key into a lock — can latch on, triggering its maturation into either a helper or killer T cell.
Helper T cells then activate immature B cells whose BCRs also conform to the shape of the presented antigen to mature them into either plasma cells that produce antibodies to remove the foreign material from the body or memory cells that “remember’ the antigen’s biochemistry for a faster response to future invasions.
Killer, or cytotoxic, T cells, on the other hand, directly attack the invaders to which they’ve been primed as a result of the immature T cell’s initial contact with the antigen.
However, when this process goes haywire with the B cells and T cells seeking out and attacking normal cells — the case of mistaken identity known as an autoimmune response — the results can be devastating.
For type 1 diabetes, scientists have long believed that the immune system somehow becomes confused and sees insulin as a target. Therefore, the misguided cellular defense forces wage war on the beta cells in the pancreas that produce the hormone, drastically lowering the amount available and leading to the high blood sugar levels characteristic of diabetes.
What isn’t well understood is the mechanism that drives the assault against the beta cells. The DE cell identified by Hamad and his colleagues — and a unique protein that it produces — appear to be the key agents for at least one possible pathway.
“It is well accepted that insulin is seen as an antigen by the T cells and that this occurs when the hormone is bound to a site on the APC known as HLA-DQ8,” Hamad explains. “However, our experiments indicate that it is a weak binding and not likely to trigger the strong immune reaction that leads to type 1 diabetes.”
Instead, the findings from the new study show that when a second protein — one coded by the BCR present on the DE cell — is substituted for insulin, it binds so tightly that it can elicit a T cell response 10,000 times stronger.
Computer simulations conducted by Ruhong Zhou, Ph.D., and his team at the IBM Thomas J. Watson Research Center were used to reveal the underlying molecular mechanism for the unusual binding of the DE cell protein, known as the x-Id peptide, and predict the strength of the T cell response to it. Additional simulations confirmed the x-Id peptide’s power, showing that it also displayed a tenfold increase in T cell activity over a laboratory-engineered “superagonist” insulin mimic that was genetically altered to create a more antigenic molecule (and is itself 1,000 times more immune stimulating than normal insulin).
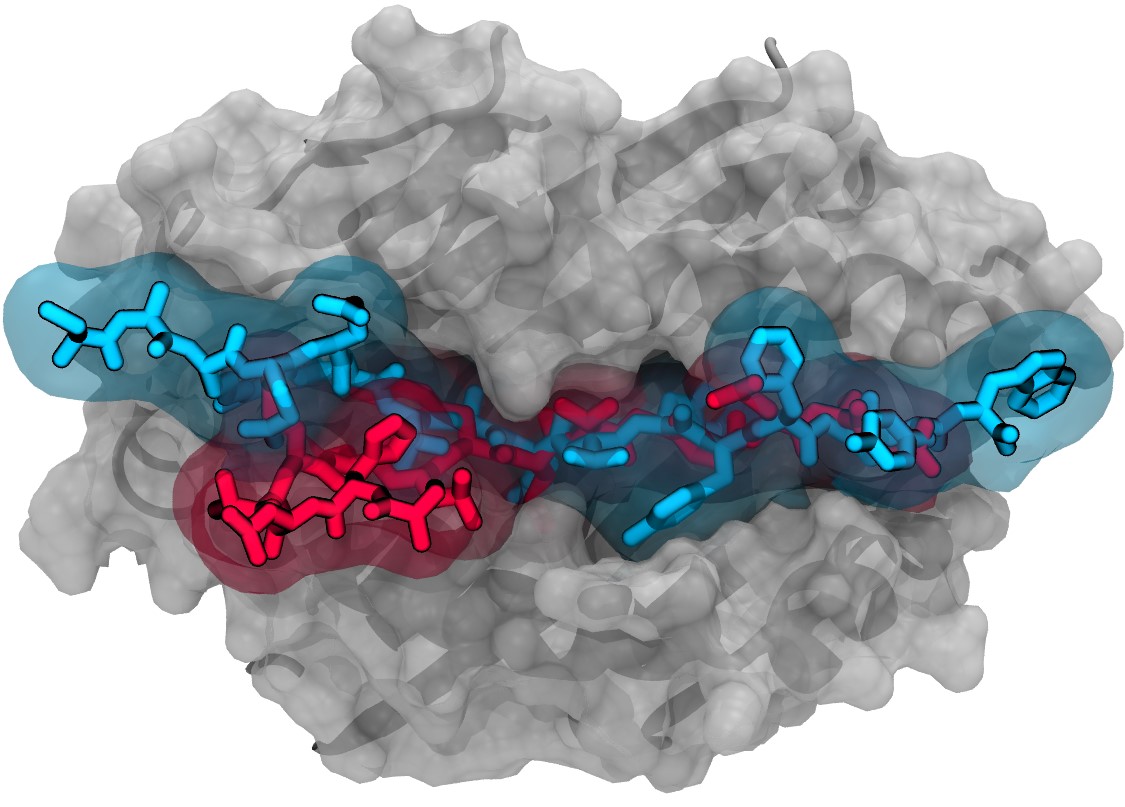 |
| Computer simulation showing the extremely tight binding to an immune system cell of a peptide (blue) produced by the newly discovered “X lymphocyte.” The protein may direct the mistaken destruction of healthy insulin-producing cells in the pancreas, and in turn, cause type 1 diabetes. This autoimmune response is 10 times stronger than that seen with a weaker-binding, lab-engineered insulin mimic (red) that is itself 1,000 times more immune stimulating than normal insulin. Credit: IBM Thomas J. Watson Research Center |
Different methods were used to verify the existence of the DE cell and define its characteristics, including modifying DE cells using a virus to give rise to a large number of DE cell clones (genetically exact duplicates). The researchers found that every clone possessed both BCRs and TCRs, proving that the lymphocyte was truly a hybrid of B and T cells.
Perhaps the most intriguing part of the “X cell story” is that the researchers found DE lymphocytes and the x-Id peptide more frequently in the blood of type 1 diabetes patients than in healthy, nondiabetic subjects. “This finding, combined with our conclusion that the x-Id peptide primes T cells to direct the attack on insulin-producing cells, strongly supports a connection between DE cells and type 1 diabetes.”
Next, Hamad says, his team will study that probable link in greater depth to confirm and more extensively define it. He says that such knowledge could lead to the development of methods to screen individuals at risk for developing type 1 diabetes.
“It also is possible that this study could lay the groundwork for developing immunotherapies that target DE cells for elimination or genetically alter the lymphocytes so that they cannot stimulate an immune response,” says Thomas Donner, M.D., director of the Johns Hopkins Diabetes Center and a co-author of the study.
Quite remarkable, Hamad says, for a “biological Nessie” that most experts told him didn’t exist.
“We were willing to take the risk and look at something different, and now we may have taken the first steps toward finding new strategies to cure type 1 diabetes,” he says. “We also may one day find that DE cells are involved in the pathology of other autoimmune disorders such as multiple sclerosis and rheumatoid arthritis.”
Besides the contributors from the Johns Hopkins University School of Medicine, the Johns Hopkins Bloomberg School of Public Health and IBM, the research team includes members from the Barbara Davis Center for Diabetes at the University of Colorado School of Medicine, Columbia University, Des Moines University and Massachusetts General Hospital.
This work was supported by grants from the National Institutes of Health (R0 AI099027) and the Norman Raab Foundation.