Link Found Between Pediatric Osteoporosis and Anti-Inflammatory Drugs
03/06/2018
By studying mice in late adolescence, Johns Hopkins University researchers have discovered that the rapid bone growth associated with puberty is slowed not only by fewer cartilage cell divisions but also by the “aging” of bone cell precursor cells. After investigating the signaling molecules that promote this transition, the scientists conclude that some weak and brittle bone conditions in both children and adults may be due to the cells’ premature “retirement” caused by glucocorticoid treatments given during puberty to treat chronic inflammation resulting from rheumatoid disorders and other diseases.
“Our results not only shed light on the dynamic cellular changes affecting bone growth during puberty, but also give us a clue as to why glucocorticoids, such as prednisolone, cause osteoporosis and bone fractures in children, and how to prevent it,” says Mei Wan, Ph.D., professor of orthopaedic surgery at the Johns Hopkins University School of Medicine.
A summary of the study was published online in the journal Nature Communications on Nov. 3.
Wan says glucocorticoids are often given to children suffering from chronic inflammation that comes with conditions such as rheumatoid disorders, acute lymphoblastic leukemia and certain genetic diseases, such as Duchene muscular dystrophy. “They work well against painful inflammation, but they also cause pediatric osteoporosis and vertebral fractures. This study brings us closer to understanding why,” she adds.
Wan says that 6 to 10 percent of children (including adolescents) who begin chronic, high-dose glucocorticoid treatment will develop osteoporosis, and 29 to 45 percent of those being treated already have it.
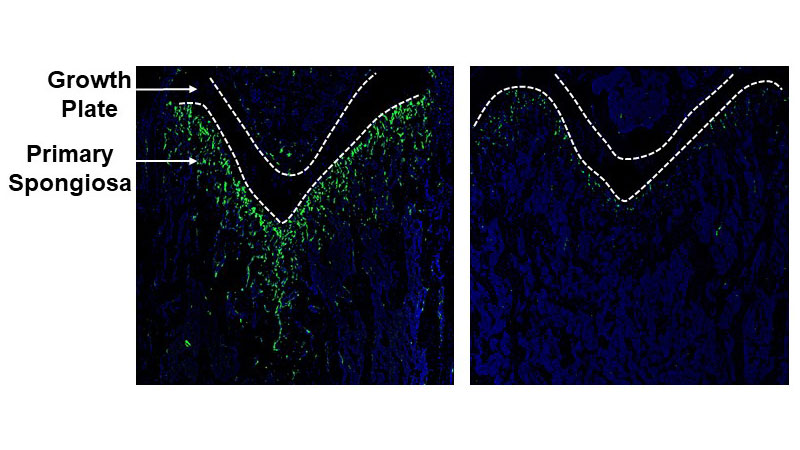
At the heart of the study, Wan says, is a region in long bones (leg and arm bones) called the primary spongiosa. It sits beneath the cartilage-rich “growth plate” at either end of each long bone. Longitudinal bone growth is primarily driven by the division and enlargement of cartilage cells in the growth plate, but the primary spongiosa provides support by housing blood vessels and mesenchymal stem cells (MSCs), which can mature into bone and cartilage cells.
In mice, as in humans, adolescence is marked by the rapid lengthening of the long bones, which tapers off in late puberty. It was already known that this rapid elongation — and its subsequent decline — is driven primarily by the division, or lack thereof, of cartilage cells in the growth plate. Whether the primary spongiosa contributed to growth rate changes in bone size and mineral accrual, however, was unknown.
Seeking answers, the research team began by looking for cellular “senescence” in the primary spongiosa of mouse femurs during puberty. Senescence is a cellular version of old age; it is commonly used by the body to control tissue growth since it halts cellular division. As expected, there were few senescent cells in early puberty (4 weeks of age) and three to four times as many in late puberty (6 and 8 weeks old). The average life span of the mice they studied is two years.
Next, researchers removed and analyzed MSCs from the femurs of mice whose MSCs had been genetically engineered to glow green when the protein nestin was produced. Nestin is used by some cells during division. Indeed, the researchers found that its presence correlated with other indicators of cell division and its absence correlated with markers of senescence.
To learn what controls MSC senescence, the scientists gave mice daily injections in early puberty of two substances known to stimulate bone buildup: growth hormone and intermittent PTH. Both treatments caused an increase in nestin-positive MSCs in late pubertal mice. However, when the glucocorticoid prednisolone was given to mice during “childhood” and early puberty (weeks two to four), the number of nestin-positive cells was significantly reduced, suggesting that the MSCs were forced into senescence earlier than usual.
Looking for the molecular signals responsible for this phenomenon, the researchers assessed the activity of 86 genes that code for proteins known to produce so-called epigenetic changes by altering the activity of other genes. There was just one enzyme, Ezh2, whose levels were significantly lower in nestin-negative MSCs compared to nestin-positive MSCs.
Ezh2 adds a molecular tag to the DNA-associated protein histone 3, which represses the activity of nearby genes. Genetic tests showed that this tag, known as H3K27me3, specifically represses the activity of senescence-inducing genes, encouraging bone growth.
While studying the effects of another enzyme, UTX, the researchers identified what could become a preventive treatment for children who need glucocorticoids. The chemical GSK-J4 enhances pro-growth, nestin-positive MSCs and bone mass by increasing H3K27me3. The researchers already have plans to test it in more mouse studies and in larger animals in the hopes that it can counteract the bone-weakening effects of glucocorticoids.
Other authors of this paper include Changjun Li, Yu Chai, Lei Wang, Bo Gao, Hao Chen, Peisong Gao, Feng-Quan Zhou, Janet Crane and Xu Cao at the Johns Hopkins University School of Medicine; Xianghang Luo at the Xiangya Hospital of Central South University, Hunan, China; and Bin Yu at the Southern Medical University, Guangdon, China.
Funding for this study was provided by the National Institute of Diabetes and Digestive and Kidney Disorders (DK083350).