Johns Hopkins Medicine Suggests Eliminating Nerve Cell Protein May Stop ALS, Dementia
07/29/2021
Amyotrophic lateral sclerosis (ALS), commonly known as “Lou Gehrig’s disease,” is a devastating neurodegenerative illness that causes nerve cells in the brain and the spinal cord to atrophy (waste away), usually resulting in dementia. Ninety percent of ALS cases are sporadic, with no known genetic mutation responsible, while the remaining 10 percent are genetically passed from parent to child. Now, Johns Hopkins Medicine researchers have identified a defective cellular pathway that initiates nerve cell breakdown and may be tied to both forms of the disease. They also suggest that eliminating charge multivesicular body protein 7 (CHMP7), the wayward protein responsible for the broken pathway, might provide a future means of treating ALS and dementia.
Led by Jeffrey Rothstein M.D., Ph.D., director of the Pedersen Brain Science Institute and the Robert Packard Center for ALS Research, and professor of neurology and neuroscience at the Johns Hopkins University School of Medicine, and senior postdoctoral fellow Alyssa Coyne, Ph.D., the research team studied CHMP7 within an area of a nerve cell known as the nuclear pore. The nuclear pore — a large, highly organized complex of different proteins — serves as the gatekeeper for the nucleus, the cell’s control center, by governing the in-and-out movement of genetic material (RNA) and proteins. Previous studies by Rothstein’s team showed that when this process goes awry — which is said to be dysregulated — it can result in the development of either the familial or inherited forms of ALS and dementias.
However, it has remained unknown why this dysregulation occurs, how it starts the chain of events leading to ALS and if it occurs in the far more common sporadic forms of ALS and dementia.
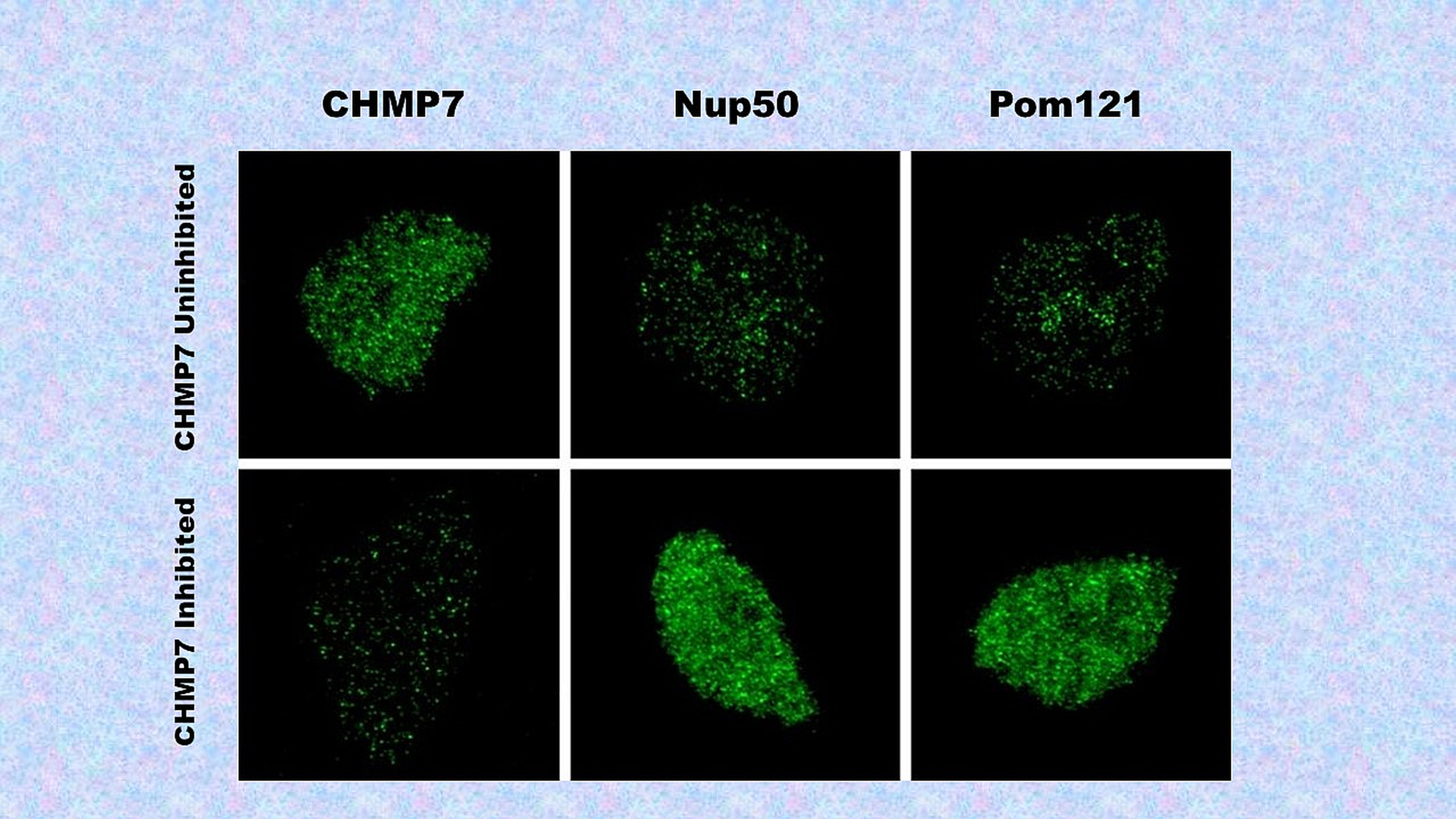
The Johns Hopkins Medicine study, published July 28, 2021, in the journal Science Translational Medicine, evaluated why the nuclear pore starts to degrade in patients with ALS and dementia, and the role of CHMP7 in that destruction.
The researchers found that CHMP7 protein accumulates specifically within the nucleus of neurons, starting a cascade of events that leads to nuclear pore injury. This, in turn, causes dysregulation of essential proteins in the cell — including one called TAR DNA binding protein 43 (TDP-43), where the dysregulated form is seen in both sporadic and familial ALS — and ultimately, cell death.
“Think of it like an engine that’s starting to lose parts,” Rothstein says. “If CHMP7 is making the nuclear pore start to degrade, what would happen if we took that insult away?”
To find the answer, Rothstein and his colleagues eliminated the CHMP7 protein with antisense technology, a new and promising tool for controlling gene expression (production of a protein) in a cell. Using synthetic antisense oligonucleotides (fragments of genetic material), the researchers targeted the CHMP7 gene at the level of messenger RNA — which codes for CHMP7 protein to be made — rather than changing the DNA — which provides the code. This inhibited the CHMP7 gene from producing its protein, and in turn, the researchers found it prevented nuclear pore degradation, subsequent cellular dysfunction and cell death. With the pore stable, they believe that the conditions potentially leading to ALS cannot be triggered.
“We’re essentially preventing the biological events that give birth to ALS,” Rothstein says.
He says the next steps in the research are to determine if this discovery can be applied to a potential ALS therapy in humans. The team also hopes to learn what causes CHMP7 to become dysregulated in patients with ALS.
Rothstein is available for interviews.