Drug Treats Inflammation Associated With Genetic Heart Disease That Can Be Deadly in Young Athletes
10/17/2019
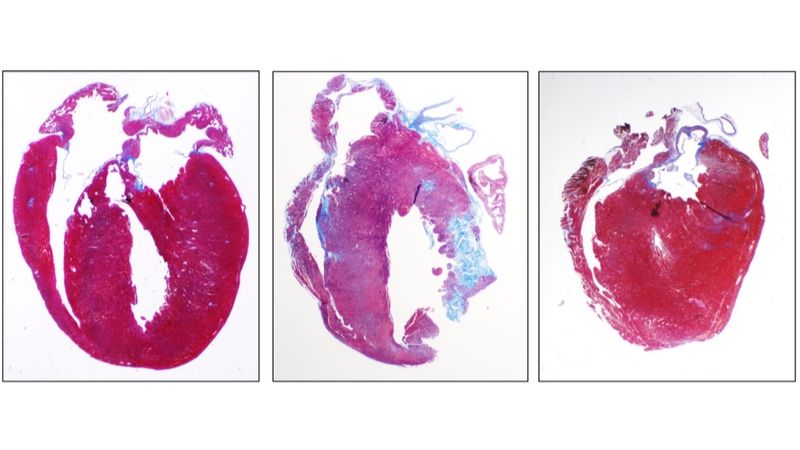
When young athletes experiences sudden cardiac death as they run down the playing field, it’s usually due to arrhythmogenic cardiomyopathy (ACM), an inherited heart disease. Now, Johns Hopkins researchers have shed new light on the role of the immune system in the progression of ACM and, in the process, discovered a new drug that might help prevent ACM disease symptoms and progression to heart failure in some patients.
“We realized that heart muscle inflammation in ACM is much more complicated than we thought, but also might provide a therapeutic strategy,” says Stephen Chelko, Ph.D., assistant professor of medicine at the Johns Hopkins University School of Medicine and senior author of the new paper, in Sept. in Circulation.
In ACM, patients often harbor mutations in any of the five genes that make up the cardiac desmosome — the gluelike material that holds heart cells together and helps coordinate mechanical and electrical synchronization of heart cells. Because of this, it’s often called “a disease of the cardiac desmosome.” In patients with ACM, heart cells pull apart over time, and these cells are replaced with damaged and inflamed scar tissue. These scars can increase risk of instances of irregular heart rhythms and lead to sudden cardiac death if the scar tissue causes the heart wall to stiffen and renders it unable to pump.
If a person is aware they carry an ACM-causing genetic mutation, doctors help them avoid cardiac death through lifestyle changes, such as exercise restriction, and medications that keep their heart rate low. However, there are currently no drugs that treat the underlying structural defects of the desmosome. People who live for many years with ACM still accumulate scar tissue and inflammation in their hearts, leading to chronic heart disease.
“We tended in the past to view ACM as something that kills due to a sudden arrhythmic event,” said Chelko. “But now we’re starting to also see it as a chronic inflammatory disease that can progress more slowly over time, leading to heart failure.”
Chelko and his colleagues wanted to determine the molecular cause of inflammation in the hearts of people with ACM. So they studied mice with an ACM-causing mutation, as well as heart muscle cells generated from stem cells isolated from an ACM patient. They found that the inflammation associated with the disease arose from two separate causes. First, they noticed high levels of macrophages, a type of immune cell that’s normally found at sites of inflammation, such as around cuts or scrapes that are healing.
“Macrophages are usually the good guys who help heal a wound and then leave,” said Chelko. “But in ACM they’re permanently setting up shop in the heart, which, over time, reduces its function.”
Chelko’s team also found that in ACM, the heart cells themselves are triggered by a protein known as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to produce chemicals called cytokines, which act as homing beacons for other inflammatory cells and molecules. When the researchers treated mice or isolated cells with a drug blocking NF-κB, heart cells stopped producing many of these cytokines, leading to decreased inflammation and infiltration of inflammatory cells. In mouse models of ACM, animals treated with the NF-κB-blocking drug Bay-11-7082 had a twofold increase in heart function, measured by how much blood their hearts could pump over time compared with untreated ACM animals. They also had a twofold reduction of damaged and inflammatory scar tissue in the heart.
More than one-third of patients with ACM who die of sudden cardiac death have no previous cardiac symptoms, so wouldn’t ever know to seek treatment. However, for relatives of these people who discover that they carry a genetic mutation causing ACM — or those who discover the mutation for other reasons — a drug could help stave off long-term heart disease, Chelko said.
While the Bay-11-7082 drug is currently only used in the lab for experimental purposes, the U.S. Food and Drug Administration has approved canakinumab, a drug that targets the same inflammatory pathway, for use in juvenile arthritis and a collection of rare auto-inflammatory syndromes. Canakinumab is also being studied for use in coronary artery disease. Chelko’s group is now investigating whether this drug would have the same effect as Bay-11-7082 in ACM.
“We’re very excited to have found an FDA-approved drug that can reduce heart inflammation in ACM, and we’re eager to do more research to ultimately help those who carry these genetic mutations,” said Chelko.
In addition to Stephen Chelko, authors on the Circulation paper are Justin Lowenthal, Djahida Bedja, Nuria Amat-Alarcon, Peter Andersen and Leslie Tung of Johns Hopkins; Angeliki Asimaki and Carlos Bueno-Beti of St. George’s University of London; Arianna Scalco of University of Padova; Daniel Judge of Medical University of South Carolina; and Jeffrey Saffitz of Beth Israel Deaconess Medical Center.
The work was supported by grants from St. Jude Medical (2015–2016 Heart Rhythm Society Cardiac Pacing and Electrophysiology Fellowship Award), The Aiding Hearts Foundation, the American Heart Association (18TPA34170559), the Gilead Research Scholars in Cardiovascular Disease Fund and the British Heart Foundation (PG/18/27/33616), Eunice Kennedy Shriver National Institute of Child Health and Human Development (1R01HD086026), TEDCO (2015-MSCRFI-1622) and the National Heart, Lung, and Blood Institute (5R01 HL120959).
Novartis Pharmaceuticals has joined Chelko’s efforts in targeting inflammation in ACM and has supplied his lab with canakinumab.