Bacteria Play Critical Role In Driving Colon Cancers
02/01/2018
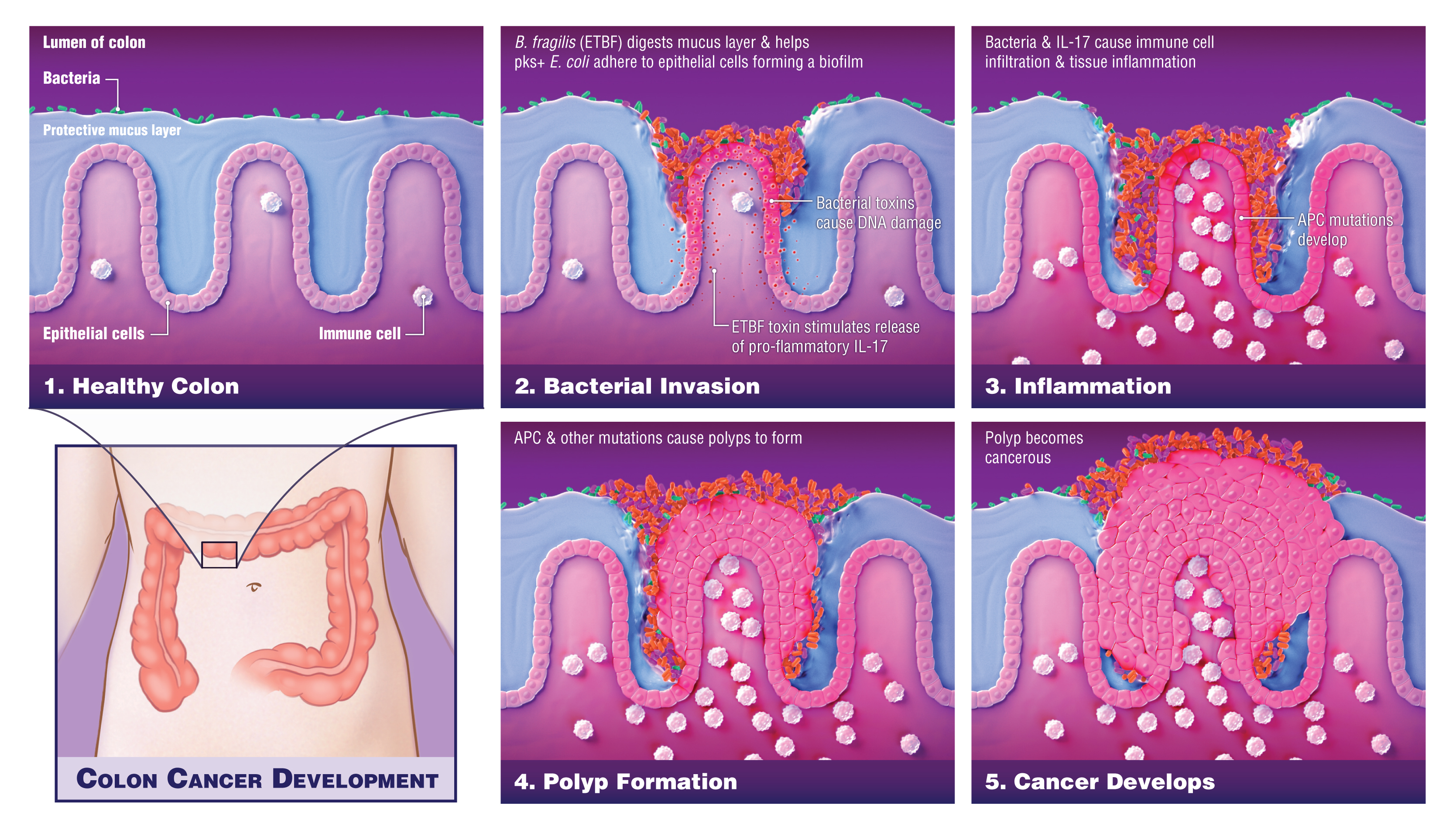
The complementary findings were published online Feb. 1 in Cell Host & Microbe and in the Feb. 2 issue of Science.
The Science findings describe a process in which these bacteria invade the protective mucus layer of the colon and collude to create a microenvironment—complete with nutrients and everything the bacteria need to survive—that induces chronic inflammation and subsequent DNA damage that supports tumor formation. These findings suggest a change in the standard of care for people who carry both types of bacteria. “More frequent colon cancer screening than the currently recommended once every 10 years should be considered,” says Drew Pardoll, M.D., Ph.D., director of the Bloomberg~Kimmel Institute for Cancer Immunotherapy. Ultimately, once better understood, administering drugs or vaccines to prevent colonization of the bacteria in the colon, and potentially even probiotics to chase the bugs from the colon, are preventive measures that could be explored to interrupt the cancer-promoting process.
These new findings by Cynthia Sears, M.D., professor of medicine, and a researcher at the Johns Hopkins Kimmel Cancer Center’s Bloomberg~Kimmel Institute, are built upon her earlier research showing that particular strains of bacteria can invade the colon mucus in at least half of patients who get colon cancer but who have no inherited predisposition for the disease. Unlike most bacteria, which do not make it past the colon’s protective mucus layer, these communities of bacteria that invade the mucus form a sticky biofilm right next to the colon epithelial cells that line the colon, where colon cancer usually originates. There, Sears and colleagues propose, these bacterial communities may eventually help the epithelial cells to become cancerous.
About 5 percent of colon cancers are caused by a hereditary syndrome called familial adenomatous polyposis (FAP), in which an inherited mutation launches a series of genetic changes that develop over time and eventually prompt the epithelial cells to turn malignant. However, Sears says, it was unclear whether ETBF or other bacteria played a role in the progression to colon cancer in FAP patients.
To investigate the relationship between the bacteria-caused biofilms and cancer formation, she and her colleagues examined colon tissue removed from six FAP patients. Tests showed patchy sections of biofilms distributed along the colon’s length in about 70 percent of the patients. The researchers used gene probes to identify the particular bacterial species and found that the biofilms consisted mainly of two types, Bacteroides fragilis and Escherichia coli, a surprising finding since the colon contains at least 500 different types of bacteria. Tests on 25 additional colon samples from FAP patients showed that the B. fragilis strain was a subtype, called ETBF, which makes a toxin that triggers certain oncogenic, or cancer-promoting, pathways in colon epithelial cells and causes colon inflammation. The E. coli strain produced a substance called colibactin (synthesized by a set of genes in the bacterial genome called the PKS island), which causes DNA mutations. “It is the combination of these effects, requiring coexistence of these two bacteria, that creates the ‘perfect storm’ to drive colon cancer development,” says Sears. Both types of bacterium are found to commonly colonize young children worldwide, potentially contributing to the rise in colon cancer rates among younger people.
“FAP is a devastating disease that ultimately results in surgical removal of the colon, and our findings could point us to new and less invasive ways to prevent colon cancer from developing,” says Sears. She says these same approaches could be applied to the more common sporadic types of colon cancer, which occur without a familial predisposition. Currently, colonoscopy to monitor for the formation of precancerous tumors, called polyps, is the standard of care. If further research shows that biofilms develop before polyps appear, Sears says, adding biofilm evaluation or stool identification of particular bacteria to care could also provide an opportunity for earlier, nonsurgical intervention that could bump the bacteria from the colon.
Using a mouse model of colon cancer, the researchers found that animals whose colons were colonized with just one of these species developed few or no tumors. However, when their colons were colonized with both species simultaneously, they developed many tumors, suggesting a synergy between the two types of bacteria.
An earlier, 2009 study, published by the Sears laboratory in Nature Medicine, suggested a unique type of immune response—producing an inflammatory protein called IL-17—was key to ETBF-induced tumor formation. Pardoll and Sears say it is important to note that this type of immune response is distinct from, and in fact antagonistic to, the types of antitumor immune responses induced by therapeutic immunotherapy drugs.
In order to prove the importance of IL-17 in the cancer-promoting effects of the bacterial combination, they used a mouse model in which the IL-17 gene was genetically deleted so it could not make IL-17, and colonized the mice with both ETBF and PKS+E. coli. Unlike animals that readily made IL-17, the genetically altered mice didn’t form colon tumors, confirming the importance of this protein in bacterial-driven colon cancer. However, in addition to IL-17, the studies showed that ETBF digested the mucus layer, enabling the PKS+ E. coli to adhere in larger numbers to the colon mucosa where together the bacteria induced increased DNA damage, a step preceding the gene mutations that underlie colon tumor formation.
The complementary findings in Cell Host & Microbe demonstrate how ETBF’s toxin prompts colon cancer to develop. Using a different mouse model of colon cancer, the researchers colonized the animals’ colons with ETBF and then performed a series of tests to monitor the resulting cellular and molecular changes.
Their results revealed that ETBF’s toxin spurs a cascade of events that promote colon inflammation that feeds back to act on the colon epithelial cells. First, the toxin triggers colon immune cells to produce IL-17. This inflammatory molecule then acts directly on the colon epithelial cells to trigger activation of a protein complex involved in promoting further inflammation, known as NFkappaB. NFkappaB in turn induces the colon epithelial cells to produce several signaling molecules that recruit more immune cells, called myeloid cells, to the colon. These immune cells are involved in the inflammatory response and are known to support tumor growth. This process culminates in tumors forming in the colon. Additional experiments showed that a protein known as STAT3, which was previously shown to play a role in regulating cancer and inflammatory genes, is also necessary for tumor formation.
Together, Sears says, the two new studies suggest a variety of strategies that researchers could test to prevent or even combat colon cancer. For example, it may be possible to prevent this disease by keeping the colon from becoming colonized by these two problematic bacteria, or by devising drugs or vaccines that target their toxins. Researchers might eventually be able to halt the malignant cascade by targeting one or several of the different molecular players that increase inflammation in the colon. The researchers are also working to better understand why the immune system permits the ongoing inflammation.
In addition to Sears and Pardoll, other researchers who participated in the Science study include Christine M. Dejea, Payam Fathi, John M. Craig, Abby L. Geis, XinQun Wu, Christina E. DeStefano Shields, Elizabeth M. Hechenbleikner, David L. Huso, Robert A. Anders, Francis M. Giardiello, Elizabeth C. Wick, Hao Wang, Shaoguang Wu, and Franck Housseau, all of Johns Hopkins, and Annemarie Boleij and Rahwa Taddese, of Radboud University Medical Center.
The Science Research was supported by the Bloomberg Philanthropies, and by National Institutes of Health grants R01 CA151393, K08 DK087856, 5T32CA126607-05, P30 DK089502, P30 CA006973 and P50 CA62924, Bristol-Myers Squibb Co.-International Immuno-Oncology Network Resource Model 300-2344, the Alexander and Margaret Stewart Trust, the American Society of Colon and Rectal Surgeons, the Netherlands Organisation for Scientific Research NWO 825.11.03 and 016.166.089, and the Institute Mérieux.The Cell Host and Microbe research was supported by the Bloomberg Philanthropies and National Institutes of Health grants R01DK080817, R01CA151325, R01GM111682, P30DK089502, P30CA006973, P50CA062924 and K08 DK087856.
COI: Pardoll discloses consultant relationships with Aduro Biotech, Amgen, AstraZeneca, Bayer, Compugen, DNAtrix, Five Prime, GlaxoSmithKline, ImmuneXcite, Jounce Therapeutics, NexImmune, Pfizer, Rock Springs Capital, Sanofi, Tizona, Janssen, Merck, Astellas, FlX Bio, Ervaxx and DNAX. Pardoll receives research support from Bristol-Myers Squibb, Compugen, Ervaxx and Potenza, and is a scientific advisory board member for Immunomic Therapeutics. He shares intellectual property with Aduro Biotech, Bristol-Myers Squibb, Compugen, and Immunomic Therapeutics. All other authors declare no competing interests. Sears, Pardoll, Dejea and Wick are inventors on patent application PCT/US2014/ 055123, submitted by The Johns Hopkins University, that covers use of biofilm formation to define risk for colon cancer. The terms of these arrangements are being managed by The Johns Hopkins University in accordance with its conflict of interest policies.