Finding A Cell’s True Identity
05/28/2019
Scientists have long sorted cells into different varieties based on their appearance under a microscope or, for differences that are more visually subtle, based on the behavior of a handful of genes. But in a bid to reveal even more distinctive differences and similarities, researchers from the Johns Hopkins Kimmel Cancer Center, the Johns Hopkins Institute for Genetic Medicine and the Johns Hopkins Department of Neuroscience developed two new artificial intelligence methods that decipher complex gene activity controlling cell fate decisions in retina development and relate this gene activity to what occurs in other tissues and across different species.
The findings, published May 22, 2019, in Cell Systems, describe commonalities and differences responsible for cellular function that can provide key insights into the study and treatment of human disease well beyond the retina.
Study leaders Elana Fertig, Ph.D., associate professor of oncology, Loyal Goff, Ph.D., assistant professor of neuroscience, and colleagues used the developing mouse retina as their model system. “The retina makes an ideal model system to study development because it mirrors the development of the entire brain, albeit with a more limited number of cell types,” says Genevieve Stein-O’Brien, Ph.D., lead author on the study.
The researchers used data derived from new single cell sequencing technology that probed which genes were turned on and to what extent—of more than 120,000 individual cells in the mouse retina over 10 developmental time points: embryonic Day 11, about halfway through gestation through postnatal Day 14 in a complementary study jointly led by Seth Blackshaw, Ph.D., professor of neuroscience, and Goff.
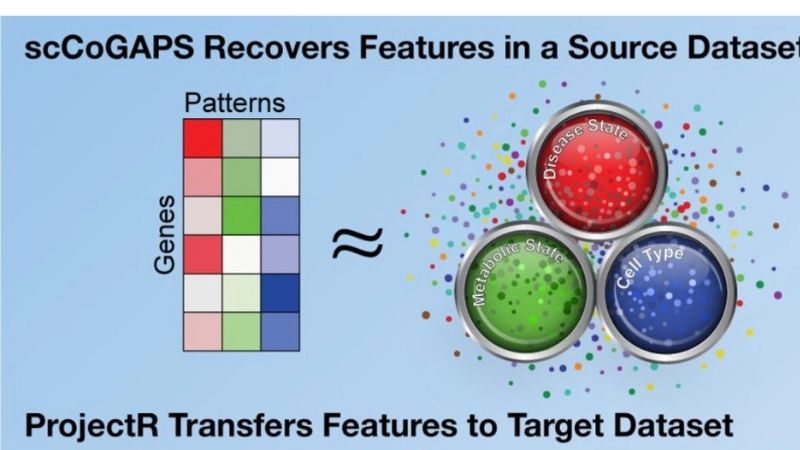
They ran this data through two different artificial intelligence programs the team had developed. The first method, called scCoGAPS, searches for patterns that characterize cells based on their expression levels. The second method, called projectR, uses these patterns to relate the cells in the developing retina to cells in other systems — including the adult mouse retina, the developing human retina, the developing brain and other cell types throughout the mouse body.
With scCoGAPS, Fertig says, the researchers identified 80 different patterns across the full developmental time course of the mouse retina. These patterns corresponded to changes from stem cells to more mature ones, discrete distinctions between mature cell types, and to differences between the mice they were derived from, such as sex, among other factors.
When the researchers projected these patterns onto cell expression data from the adult mouse retina, they found clear similarities — for example, patterns for gene expression in adult rod cells in the retina shared features with cells in the developing mouse retina that were maturing into rods. However, most patterns of gene expression differed between the two sets, with the embryonic and postnatal cells expressing patterns that were clearly developmental in nature that contrasted with those of the mature cells. As an example, Stein-O’Brien explains “the developmental patterns learned with CoGAPS reveal changes in gene expression and cell cycle that cause a common progenitor to give rise to totally different cell types.”
The researchers also saw common patterns between the developing mouse retina, the developing human retina, the developing human cerebral cortex and the developing mouse midbrain. These similarities show that the artificial intelligence programs can be useful for making comparisons between developmental points and between different species and organ systems, explains Fertig.
Delving deeper, the scientists compared the patterns in developing mouse retina cells with gene expression in cells elsewhere in the mouse body. Surprisingly, Fertig says, they found similar gene expression between secretory cells in the lungs and a subset of cells in the retina, hinting at the presence of an as-yet undescribed cell type in the lung based on what was learned in the retina.
“From a basic biology standpoint, this challenges the notion that a given cell type is really defined by a limited set of genes,” she says. “We show that these patterns of genes can be interrelated and that broader patterns across genes are preserved from sample to sample and context to context.”
These artificial intelligence tools could also shed light specifically on cancer, Fertig adds, the main focus of her research overall. For example, some tests aim to gauge patients’ response to cancer therapies by the activity of a single gene or a handful of genes. Using scCoGAPS and projectR may show a more complicated picture of gene activity that could help researchers develop better ways to target cancers.
Other researchers who participated in this study include Brian S. Clark, Thomas Sherman, Cristina Zibetti, Sheng Liu, Jian Qian, Carlo Colantuoni, Seth Blackshaw and Loyal A. Goff, all from Johns Hopkins; Qiwen Hu, from the University of Pennsylvania; and Rachel Sealfon, from the Flatiron Institute.
This work was supported by grants from the NIH (R01EY020560 and U01EY027267, F32EY024201 and K99EY027844, R01CA177669, U01CA196390, U01CA212007, and P30CA006973), the NSF (IOS-1656592), the Chan-Zuckerberg Initiative DAF (2018-182718, 2018-183445, and 2018-183444), an advised fund of Silicon Valley Community Foundation, the Johns Hopkins University Catalyst and Discovery awards, and the Johns Hopkins University School of Medicine Synergy Award.