Tau Interferes with Nuclear Transport in Alzheimer’s Disease
09/05/2018
A team of scientists led by researchers from Johns Hopkins Medicine and from Massachusetts General Hospital have found that the nuclear pore complex, which controls the transport of molecules into the cell nucleus, is defective in animal and human Alzheimer’s disease (AD) cells and that the defect is associated with aggregation of proteins in neurons.
Researchers have long known that tau, a protein that accumulates in the brains of individuals with Alzheimer’s, is a major component of AD’s hallmark neurofibrillary tangles that are presumed to be involved in the development of dementia. Precisely how tau contributes to the disease has remained a mystery. But new research publishing September 5 in the journal Neuron suggests the possibility of a new mechanism of neuronal dysfunction where tau interferes with the nucleus’s ability to communicate with the rest of the cell.
In a cell, the nucleus is surrounded by a membrane separating contents inside the nucleus from everything else within the cell. The nucleus communicates with the cell through protein-rich structures known as nuclear pores. Defects in these pores have been suggested in other causes of dementia, particularly frontotemporal dementia, and in amyotrophic lateral sclerosis (ALS).
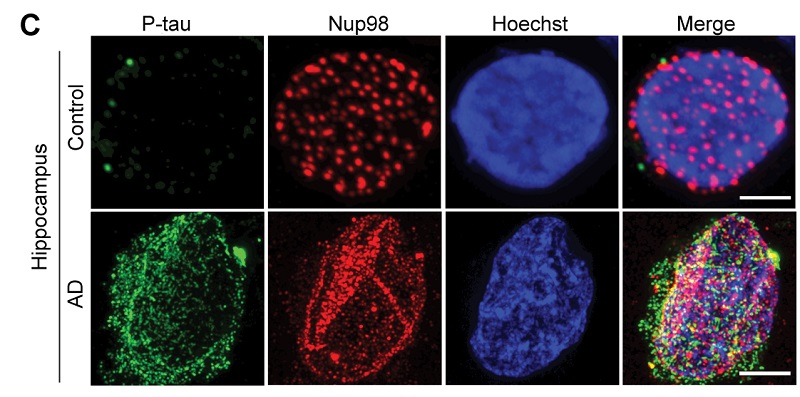
The nuclear pore complex includes more than 400 different proteins. In this research, the team focused on one of the major structural proteins of the pore, Nup98. The researchers found that in the presence of tau, the Nup98 nuclear pores are not evenly spaced throughout the structure as expected. Instead, they were physically disrupted, fewer in number, and coalesced with each other. The team also noted that Nup98 seemed to leak or be mislocalized in the cytoplasm of AD brain cells rather than remaining in the nuclear pore. Whenever it was mislocalized, those same cells tended to have aggregates of tau.
Studying human AD brain tissue, the team found that the more extreme the AD disease was while patients were alive, at autopsy they had worse pathology related to Nup98 mislocalization with tau. In mice models, when human tau was added to cultures of living rodent neurons, Nup98 was mislocalized in the cytoplasm and functional nuclear import was disrupted. “This was a functional link between tau and damage to the nuclear transport in the nuclear membrane,” says co-senior author Jeffrey Rothstein, M.D., Ph.D., professor of neurology at the Johns Hopkins University School of Medicine.
The researchers suggest that this physical alteration of the Nup98 nuclear pore in the presence of tau disrupts the function of the nuclear pore and accelerates tau aggregation and the formation of neurofibrillary tangles, ultimately leading to neural dysfunction and death. It remains unknown whether the Nup98/tau interaction is a normal function that is exaggerated in neurodegenerative disease or whether it occurs only in neurodegenerative disease.
This research was supported by the National Institute on Neurological Disorders and Stroke (P01NS099114, R01NS094239,R01NS099320, U54NS100693, R35NS097273, P01NS084974, R01NS089544 and R21NS094489), the National Institute on Aging (P50AG016574 and R01AG056306), the Massachusetts Alzheimer’s Disease Research Center, the JPB Foundation, the Swiss National Science Foundation, the Paul G Allen Family Foundation, the Glenn Foundation Center for Aging Research, the American Federation for Aging Research, the Leona M and Harry B Helmsley Foundation Charitable Trust, Annette Merle-Smith, and the Mathers Foundation.
COI: Eftekharzadeh is employed by Biogen. Daigle is employed by Abbvie.
*A version of this press release was issued by Neuron.