Cancer Risk Associated With Key Epigenetic Changes Occurring Through Normal Aging Process
02/22/2018
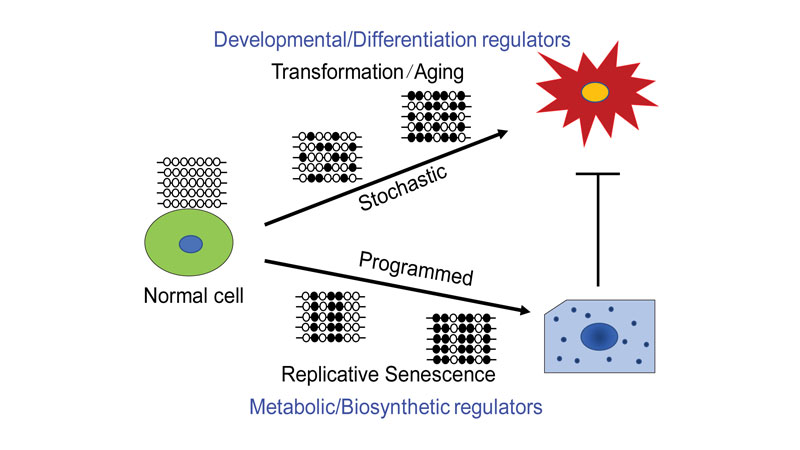
Some scientists have hypothesized that tumor-promoting changes in cells during cancer development—particularly an epigenetic change involving DNA methylation—arise from rogue cells escaping a natural cell deterioration process called senescence. Now, researchers at the Johns Hopkins Kimmel Cancer Center demonstrated that instead, tumor-associated epigenetic states evolve erratically during early stages of tumor development, eventually selecting for a subset of genes that undergo the most changes during normal aging and in early tumor development.
The work, published in the Feb. 12 issue of Cancer Cell,may help refine biomarkers of cancer risk. Screening for these genes may help stratify, for every age group, individuals with the highest risk for cancer development, the authors say.
“Aging is probably the leading risk factor for most common cancers,” says coauthor Stephen Baylin, M.D., the Virginia and Daniel K. Ludwig Professor of Cancer Research at the Kimmel Cancer Center. As epigenetic changes are reversible in laboratory models, some scientists are considering the possibility of modifying this risk factor as a potential way to modulate aging as a cancer risk factor. “Maybe you won’t find the Ponce de León Fountain of Youth, but if you could dampen down the risk of cancer for every decade you age, that could be huge,” says Baylin.
For the study, investigators studied patterns of DNA methylation, a process by which cells add tiny methyl chemical groups to a beginning region of a gene’s DNA sequence, often silencing the gene’s activation. They took fibroblast cells from human foreskin samples and followed an established, three-step laboratory process called Weinberg’s classical transformation system to convert the cells to cancer cells. The process includes infecting cells with genes known to induce tumor development. The scientists also injected the early transformed cells into mice to monitor their progression. They took a second group of fibroblast cells and let them naturally mature into senescence. The researchers followed methylation changes observed in both groups of cells over time.
“Senescence is a very well-known, normal aging process that is actually an antitumor mechanism. It occurs when cells perceive an excess of DNA damage, when cells undergo too many cell divisions or when they experience cancer development-related stress. So it was puzzling to us how the senescence process could lead to the formation of tumors,” says senior study leader Hariharan Easwaran, Ph.D., assistant professor of oncology at the Johns Hopkins Kimmel Cancer Center.
To solve the mystery, Easwaran, Baylin and collaborators began mapping the evolution of epigenetic DNA methylation alterations during senescence and transformation. They found that although the alterations appeared similar between senescent cells and transformed cells, the way they evolved and the genes that got methylated were different for the most part.
The researchers found that DNA methylation that evolved during senescence was very programmed, so much so that replicates of the senescence process in different petri dishes all had the same epigenetic pattern, Easwaran says. Independent replicates of the transformation process, by contrast, appeared very stochastic, or random.
In addition, the transformation process primarily involved regulator genes that are critical for cancers to maintain the characteristics of a cancer cell, Baylin says. In comparison, senescence involves a number of metabolic process genes.
In additional experiments, Wenbing Xie, Ph.D., the study’s lead author, could not coax senescent cells into becoming transformed cells, serving as a protection against cancer development, and identified a subset of transformation-associated methylated genes that were most prone to get methylated during early tumor formation and during aging. In future work, the researchers will explore tissue-specific patterns of DNA methylation gains for the senescence and transformation classes of genes, and hope to use that to devise strategies for determining age-associated risk-stratification of tumor development.
In addition to Baylin, Easwaran and Xie, scientists who contributed to the study were Ioannis Kagiampakis, Yang W. Zhang, Lauren Murphy, Yong Tao, Xiangqian Kong, Byunghak Kang, Limin Xia, Filipe L.F. Carvalho, Ray-Whay Chiu Yen, Cynthia A. Zahnow and Nita Ahuja of Johns Hopkins; Lixia Pan of the National Cancer Institute; and Subhojit Sen of Mumbai University in India.
The work was supported by the National Cancer Institute grants R01CA185357, R21CA212495 and R01CA170550, The Hodson Trust and the Evelyn Grollman Glick Scholar Award.