Study Finds COVID-19 Pandemic Led to Some, But Not Many, Developmental Milestone Delays in Infants and Young Children
Read Full ArticleLatest News
-
05/16/2024
Study Suggests High-Frequency Electrical ‘Noise’ Results in Congenital Night Blindness

-
05/13/2024
New Study Shows Certain Combinations of Antiviral Proteins Are Responsible for Lupus Symptoms and Affect Treatment Outcomes

-
05/13/2024
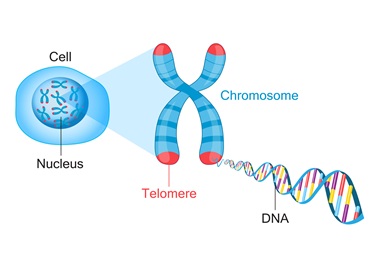
Variations In Telomere Lengthening Genes May Predispose Some People to Papillary Thyroid Cancer
Contact Johns Hopkins Media Relations
Johns Hopkins Medicine In the News
-
Feeling awe and wonder can be good for your mental health
"Nature and interpersonal experiences tend to be the most frequently listed sources of awe, but there's a lot of variation," says Sean Goldy, a postdoctoral research fellow who studies awe at the Johns Hopkins Center for Psychedelic and Consciousness Research.
-
Will I get seasick on a cruise? Here's what travelers should know.
Seasickness is a form of motion sickness. That happens when there is a difference between the information you get from your visual system, your inner ear and receptors in your muscles, according to Dr. Kathleen Cullen, a professor of biomedical engineering at Johns Hopkins University.