Brain Cell Death in ALS, Dementia Tied to Loss of Key Biochemical Transport Structure in Nucleus
09/02/2020
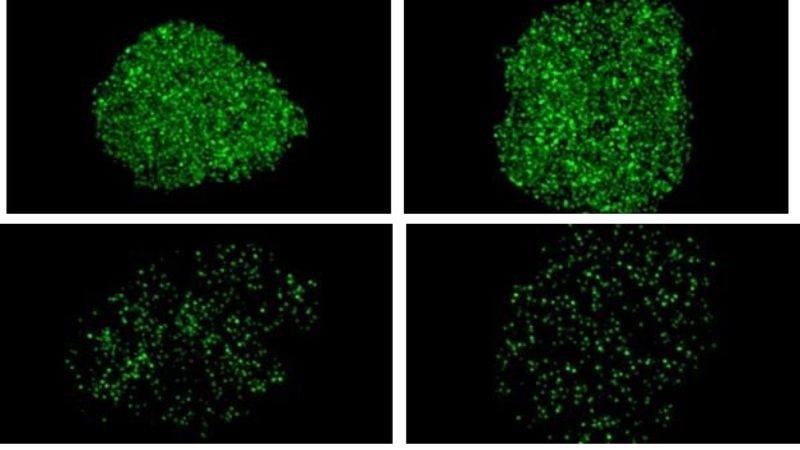
Researchers have long sought to explain precisely how the most common genetic mutation linked to both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia causes the death of nerve cells. Now, Johns Hopkins Medicine researchers report new evidence that the mutation slowly disrupts the vital transport system of proteins, enzymes and other material in and out of the cell’s control center, a process that eventually kills neurons in the brain and spinal cord.
The researchers say their findings, described online on July 15 in Neuron, confirm the pathway as a target for developing new treatments for ALS and the second-most common type of dementia. ALS, which affects an estimated 30,000 Americans, is a wasting disease in which neurons, involved in the control of muscle and motion, die. People with the disorder slowly lose the ability to move, swallow and breathe. Frontotemporal dementia, estimated to afflict up to 60,000 Americans, is marked by symptoms similar to Alzheimer’s disease, robbing people of memory, learning ability, personality and stable moods. There are no established treatments for either disease. The genetic mutation C9orf72, called C9 for short, is found in about 40–50% of people with hereditary ALS and about 12% of people with hereditary frontotemporal dementia.
“The way we think this common mutation works is like a first scratch on a new car that, if let go over time, rusts the whole car until it won’t work,” says Jeffrey Rothstein, M.D., Ph.D., director of the Brain Science Institute and the Robert Packard Center for ALS Research and professor of neurology at the Johns Hopkins University School of Medicine.
Past work from Rothstein’s lab demonstrated that a common mechanism in many neurodegenerative diseases, including ALS, is disrupted transport of enzymes, RNA and proteins in and out of the nerve cell nuclei — the control centers containing the genetic code. When this transport is blocked, the neurons die over time. These small molecules move through thousands of pores in the nucleus, dubbed nuclear pore complexes, each made of 30 different kinds of protein building blocks, called nucleoporins.
In a bid to further understand the process, the new study, led by Alyssa Coyne, Ph.D., a postdoctoral research fellow in Rothstein’s lab, added tiny chemical tags to 23 of the 30 nucleoporin proteins, which enabled them to see the nuclei’s pores using super-resolution microscopy.
They then compared the pore traffic in healthy neurons with neurons from people with the C9 ALS/dementia-causing mutation. In the C9 neurons, the researchers say they consistently saw much lower levels of eight different nucleoporins, suggesting that the pore was missing these nucleoporin “bits and pieces,” leading to disrupted transport routes in the latter cells.
Using standard tools of cell engineering, the researchers added back each one of the eight nucleoporin genes to produce higher levels of nucleoporin proteins, thereby “rebuilding” the pore in the C9 cells with ALS/dementia-causing mutation. They found that adding back the gene that encodes Pom121 nucleoporin restored levels of all seven other nucleoporins too, and restored transport of cell materials in and out of the nuclei.
“Normally, when you subject C9 neurons to stress, they die. However, when we added back the missing Pom121, the pore was ‘rebuilt,’ and the C9 neurons didn’t die when stressed,” Rothstein says. According to the researchers, Pom121 is a structural protein that links the inner and outer rings of the nuclear pores and anchors them in the nuclear membrane.
In what way does the C9 ALS/dementia mutation cause this injury to the nuclear pore? The C9 ALS/dementia mutation is caused by a repeated sequence in the DNA letters GGGGCC. This DNA sequence converts into RNA that makes RNA “repeats,” an abnormal process that issues instructions for several different proteins that also have repeated protein sequences. Researchers haven’t been certain whether the RNA repeats or the protein repeats lead to the later progression of disease, Rothstein notes.
To see if the RNA or protein repeats in the neurons with the C9 ALS/dementia-causing mutation leads the nuclear pores to fall apart, the researchers examined a type of neuron that had the entire C9orf72 DNA sequence removed. Without this DNA sequence, the neurons can’t make the RNA or protein repeats on their own. They then added in various combinations of the protein repeats, and although these protein repeats clumped in the cells, they found that the protein repeats didn’t affect the nucleoporin protein levels. Separately, they added the RNA repeats in these neurons without the C9 DNA and found the same reduced nucleoporin proteins as in neurons from ALS/dementia patients with the C9 mutation.
“This finding confirmed that drugs targeting the RNA repeats in people with the C9 mutation may be a way to treat patients with ALS or frontotemporal dementia by interfering with the ‘rust’ process,” says Coyne. “What is promising is that there are several drugs in clinical trials that target the RNA repeats by binding to them and breaking them down as a treatment for ALS.”
Additional authors on the study include Benjamin Zaepfel, Lindsey Hayes and Kelly Bowen of Johns Hopkins; Boris Fitchman, Yuval Salzberg and Amnon Harel of Bar-Ilan University; En-Ching Luo, Stefan Aigner and Gene Yeo of University of California San Diego; Hannah Trost, Clive Svendsen and Dhruv Sareen of Cedars-Sinai Medical Center; and Frank Rigo of Ionis Pharmaceuticals.
This work was supported by the ALS Association Milton Safenowitz Postdoctoral Fellowship, the National Institute of Neurological Disorders and Stroke (RF1AG062171,P01NS099114 and R01NS094239), the Robert Packard Center for ALS Research Answer ALS Program, ALS Finding a Cure, ALS Association, Muscular Dystrophy Association and the Chan Zuckerberg Initiative.
Rothstein is participating in a clinical trial for new ALS treatments from Amylyx Pharmaceuticals.