The mission of the Institute for Basic Biomedical Sciences at the Johns Hopkins School of Medicine is to promote the fundamental research that drives advances in medicine. By fostering a unique and collaborative environment that bridges basic science and clinical research, the IBBS supports and encourages interdisciplinary interactions that lead to discovery and innovation and educates and trains future leaders in biomedical research.
-
Donate to Basic Research
Philanthropic support has never been more critically important than it is today, nor has the potential return on investment been greater.
-
View Institute Events
Please check this page frequently for information about IBBS seminars and major IBBS symposia.
One Year As Department Director, Decades As Leading Cancer Metastasis Researcher
Cell biologist Andrew Ewald is known for his discoveries in how breast cancer cells spread through the body at the cellular level. With a year as department director under his belt, we spoke with Ewald about his first year and the future of cancer metastasis research.

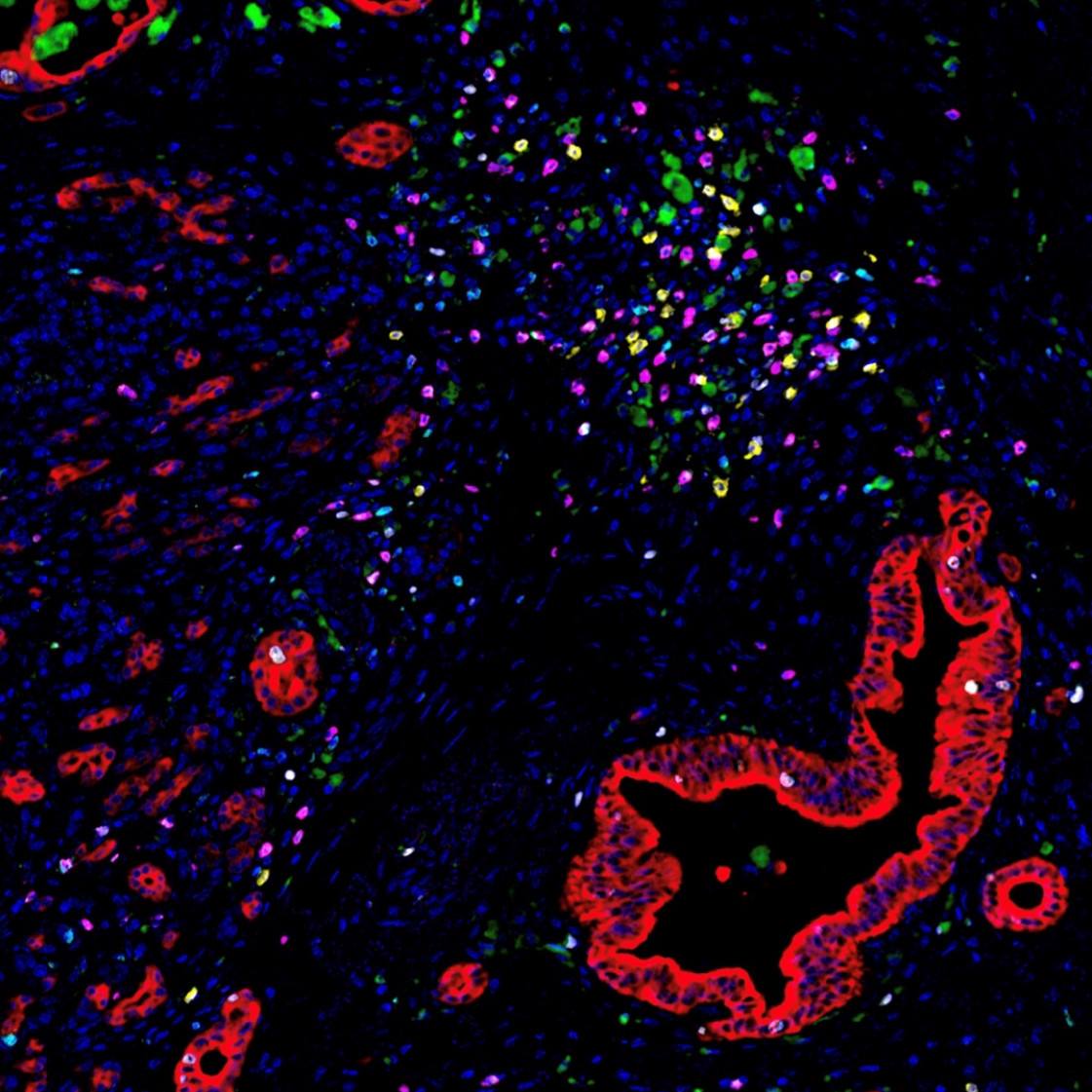
Image of the Month: The Deadliest Cancer
Pancreatic cancer is the deadliest major cancer type, but Johns Hopkins Medicine biomedical engineers discovered a way to predict how likely a patient with pancreatic cancer will respond to treatment and survive.
Johns Hopkins Researchers Build a "Bridge" to AI Technologies by Joining New NIH Consortium
Researchers are designing a system to ethically collect and generate a data set with various types of information that will be useful for many generations of scientists who specialize in using machine learning to solve challenging issues in human health.


-
Basic Science Departments
Among nine basic science departments, more than 150 make discoveries to transform medicine.

-
Core Services
The Institute for Basic Biomedical Sciences is home to several centralized fee-for-service facilities open to all researchers at Johns Hopkins and beyond.

-
Research Centers
Several research centers are supported by the Institute for Basic Biomedical Sciences.

-
Meet our Faculty
Learn about the research of some of the 150 faculty members affiliated with the Institute for Basic Biomedical Sciences.

The Beckman Center for CyroEM at Johns Hopkins
The Beckman Center for Cryo-EM at Johns Hopkins provides cutting edge cryoelectron microscopy to the Johns Hopkins and regional communities. Hear about how cryoEM is being used at Johns Hopkins from leading scientists.
Learn more about the center
Announcements
Professorships for Basic Science
Two basic science faculty members were recently honored with professorships:
Deborah Andrew was installed as the Bayard Halsted Professor in the Department of Cell Biology.
Cynthia Wolberger received the Brown Advisory Colleagues Professorship in Scientific Innovation.
Developmental Biology Accolades
Developmental biologist Deborah Andrew won this year’s Lifetime Achievement Award from the Society for Developmental Biology and the George W. Beadle Award from the Genetics Society of America for contributions to student education, faculty development and service to the Drosophila research community. Andrew studies how “tube” organs (salivary glands, trachea) form in a fruit fly, which may give insights to human development and disease.