Featured Story Telemedicine and Cardiac Rehabilitation
Research and other work to strengthen access to and ease use of cardiac rehabilitation at Johns Hopkins accelerated with the COVID-19 pandemic.
News for Physicians from Johns Hopkins Medicine
Research and other work to strengthen access to and ease use of cardiac rehabilitation at Johns Hopkins accelerated with the COVID-19 pandemic.

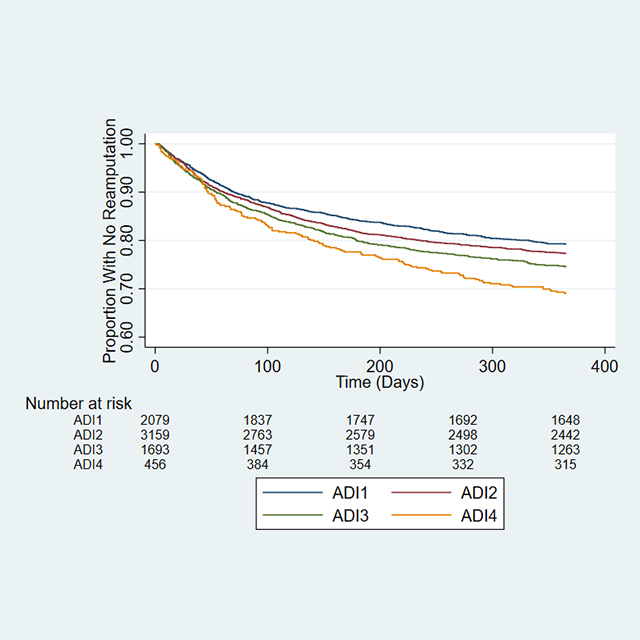
Multidisciplinary, preventive care may offset risk of amputation for certain high-risk patients.
The team at the Johns Hopkins Women’s Cardiovascular Health Center provides complete care for women with heart conditions that can affect them across their life spans.

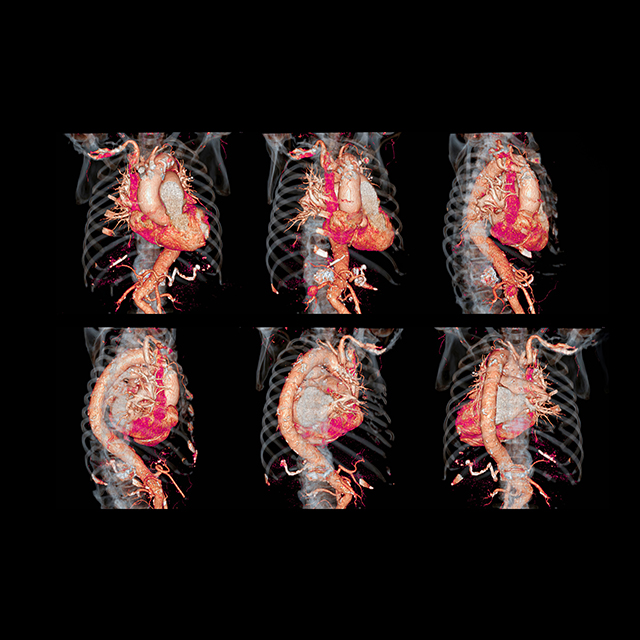
The multidisciplinary care team at Johns Hopkins’ Broccoli Center cares for the entire range of conditions that affect the largest artery.
Johns Hopkins research in animals suggests that giving patients the hypertension drug diazoxide along with cardioplegia during cardiac surgery may lessen the detrimental effects of reduced blood flow during and after procedures.
